





Sphingolipidose
Die Sphingolipidosen sind genetisch bedingte Störungen, bei denen sich fettähnliche Substanzen (Lipide) in den Zellen und bestimmten Organen anreichern.
Die Cerebroside-Speicherkrankheit ist die häufigste der Lysosomalen Speicherkrankheiten. Sie stellt eine Form der Sphingolipidose dar, da bei einer Störung des Sphingolipid-Stoffwechsels ein Defekt vorliegt.
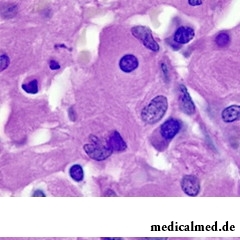 Die Zerebrosidspeicherkrankheit ist am meisten verbreitet lisossomnych der Speicherungskrankheiten. Sie ist eine der Formen sphingolipidosis (die Unterabteilung lisossomnych der Speicherungskrankheiten), da in disfunkzionalnosti des Metabolismus sfingolipidow gezeigt wird.
Die Zerebrosidspeicherkrankheit ist am meisten verbreitet lisossomnych der Speicherungskrankheiten. Sie ist eine der Formen sphingolipidosis (die Unterabteilung lisossomnych der Speicherungskrankheiten), da in disfunkzionalnosti des Metabolismus sfingolipidow gezeigt wird.
Das Krankheitsbild wird durch Milz- und Lebervergrößerung, Anämie, Thrombozytopenie sowie neurologische Komplikationen charakterisiert. Die Ursache liegt in einem erblichen Defekt des Enzyms Glukozerebrosidase, das für den Abbau von Glukosylceramid verantwortlich ist. Bei Fehlen dieses Enzyms kommt es zur Ansammlung von Glukosylceramid, insbesondere in Makrophagen (eine Untergruppe der Leukozyten). Eine Akkumulation kann zudem in Milz, Leber, Nieren, Gehirn und Knochenmark auftreten.
Die klinischen Manifestationen umfassen Splenomegalie, Hepatomegalie, schwere neurologische Komplikationen, Lymphadenopathie, Bauchauftreibung, eine bräunliche Hautverfärbung (Hepatosiderose), Anämie und Thrombozytopenie.
Die Erkrankung wird durch eine rezessive Mutation in einem Gen auf Chromosom 1 verursacht und betrifft sowohl Männer als auch Frauen. Etwa 1 von 100 Menschen weltweit ist Träger der Sphingolipidose. Die Krankheit wurde zu Ehren des französischen Arztes Philippe Gosset benannt, der sie 1882 erstmals beschrieb.
Typen der Sphingolipidosen
Die Sphingolipidose umfasst drei klinische Haupttypen: Typ I, Typ II und Typ III.
Typ I ist die häufigste Form der Erkrankung mit einer Häufigkeit von etwa 1 zu 50.000 Neugeborenen. Die Symptome können im frühen Kindesalter oder im Erwachsenenalter auftreten und umfassen:
- Lebervergrößerung sowie eine ausgeprägte Milzvergrößerung;
- Schwäche des Skelettsystems;
- Anämie, Thrombozytopenie sowie das Felty-Syndrom;
- Niereninsuffizienz;
- Erschöpfungszustände.
Der Typ II manifestiert sich gewöhnlich innerhalb der ersten sechs Lebensmonate mit einer Häufigkeit von etwa 1:100.000 Lebendgeborenen. Die Symptome dieser Form der Zerebrosidspeicherkrankheit umfassen:
- Lebervergrößerung und Milzvergrößerung;
- Ausgedehnte und fortschreitende Hirnschäden;
- Bewegungsstörungen im Auge sowie Muskelkrämpfe und Muskelsteifheit der Gliedmaßen;
- Verminderte Saug- und Schluckfähigkeit.
Betroffene Kinder sterben in der Regel im Alter von zwei Jahren.
Der Typ III (die langdauernde neuropathische Form) kann jederzeit beginnen – bereits in der Kindheit oder sogar im Erwachsenenalter – und tritt mit einer Häufigkeit von 1 auf 100.000 Neugeborene auf. Die Hauptsymptome umfassen Milz- oder Leberschwellung, Krämpfe, Koordinationsstörungen, Atembeschwerden, Skelettanomalien sowie Blutkrankheiten, einschließlich Anämie.
Symptome der Zerebroside-Speicherkrankheit
Die allgemeinen Symptome der Zerebroside-Speicherkrankheit sind:
- Eine schmerzlose Hepatomegalie und Splenomegalie können Leber- bzw. Milzgröße auf 1.500 bis 3.000 ml vergrößern, im Unterschied zum normalen Umfang von 50–200 ml. Die Splenomegalie kann den Appetit verringern, Druck auf den Bauch ausüben und das Risiko eines Milzbruchs erhöhen;
- Hyper- und Pancytopenie – die schnelle und vorzeitige Zerstörung der Blutzellen, was zu Anämie, Neutropenie, dem Felty-Syndrom sowie Thrombozytopenie führt (mit erhöhtem Risiko für Infektionen und Blutungen);
- Leberzirrhose
- Stark ausgeprägte Gelenk- und Knochenbeschwerden, insbesondere an den unteren Extremitäten und in den Kniegelenken.
- Neurologische Symptome
- Typ II: Schwere Krämpfe, Bluthochdruck, geistige Entwicklungsrückstände sowie Atemstillstand.
- Typ III: Muskelzuckungen, Krämpfe, geistige Behinderung sowie Apraxie der Augenmuskulatur
- Osteoporose
- Gelbbraune Hautpigmentierung
Therapie der Zerebroside-Speicherkrankheit
Die Therapie der Zerebroside-Speicherkrankheit (Untertypen 1 und 3) kann mit dem intravenösen Ersatz des rekombinanten Glukozerebrosidase-Ferments beginnen. Dies kann die Volumina von Leber und Milz signifikant verringern sowie Skelettanomalien adressieren; andere klinische Erscheinungsformen bleiben jedoch unbehandelt. Diese Prozedur kostet pro Patient 200.000 USD und sollte jährlich während des gesamten Lebens des Patienten wiederholt werden. Alternativ kann die Behandlung der Zerebroside-Speicherkrankheit mit dem Medikament Velaglucerase Alfa erfolgen, das seit Februar 2010 als Therapeutikum zugelassen ist.
 zurück andere Erscheinungsformen zu wenden. Diese Prozedur kostet daneben 200000$ für einen Patienten und soll jährlich während des ganzen Lebens des Kranken wiederholt werden. Auch verwirklicht sich die Behandlung der Zerebrosidspeicherkrankheit mit Hilfe des Medikaments Velaglucerase Alfa, das als Alternativbehandlung seit Februar 2010 gebilligt war.
zurück andere Erscheinungsformen zu wenden. Diese Prozedur kostet daneben 200000$ für einen Patienten und soll jährlich während des ganzen Lebens des Kranken wiederholt werden. Auch verwirklicht sich die Behandlung der Zerebrosidspeicherkrankheit mit Hilfe des Medikaments Velaglucerase Alfa, das als Alternativbehandlung seit Februar 2010 gebilligt war.
Eine weitere Therapieoption für die Zerebroside-Speicherkrankheit stellt eine erfolgreiche Knochenmarktransplantation dar. Diese behandelt jedoch nicht die neurologischen Manifestationen der Erkrankung, da während des Eingriffs Monozyten mit florider Beta-Glukosidase appliziert werden. Aufgrund des hohen Risikos und der geringen Erfolgsaussicht ist diese Maßnahme bei Zerebroside-Speicherkrankheiten selten empfehlenswert.
Die chirurgische Splenektomie wird nur in Ausnahmefällen durchgeführt, wenn eine Anämie vorliegt oder das vergrößerte Milzorgan die Gesundheit des Patienten beeinträchtigt. Bluttransfusionen können bei Patientinnen mit Anämiemerkmale indiziert sein. Zudem kann der chirurgische Gelenkersatz zur Verbesserung von Mobilität und Lebensqualität in bestimmten Fällen erforderlich sein.
Zusätzliche Behandlungsmethoden der Zerebrosidose umfassen die Gabe von Antibiotika bei Infektionen, Antiepileptika sowie Bisphosphonate zur Behandlung von Knocheninfektionen, ferner Lebertransplantationen.
Die Behandlung der Sphingolipidose erfolgt auch durch orale Gabe von Wirkstoffen, die auf molekularer Ebene wirken. Miglustat ist ein solches Präparat und wurde 2003 für die Therapie dieser Erkrankungen zugelassen.
Intellektuell aktive Menschen sind weniger anfällig für Gehirnerkrankungen; ihre geistige Tätigkeit bildet eine zusätzliche Reserve, die kompensatorisch wirkt.

Die Rolle des Immunsystems für das Kindeswachstum ist unschätzbar; vom Immunsystem produzierte IgE-Antikörper schützen das Kind vor Krankheiten...
Abteilung: Artikel über Gesundheit.
Für niemanden ist es ein Geheimnis, dass unser Land eines der alkoholstärksten Länder der Welt ist; bei klarem Verständnis davon, dass der Konsum von Spirituosen außerordentlich schädlich ist, verhalten sich die Mehrheit der Bevölkerung gegenüber dem Alkoholmissbrauch mit ungerechtfertigter Loyalität. Gerade dieser...
Abteilung: Artikel über Gesundheit.
Eine unzureichend ausgeprägte sexuelle Sucht oder das Fehlen sexueller Befriedigung kann von Zeit zu Zeit jeden Menschen betreffen; jedoch, wenn dies regelmäßig vorkommt, ist es ein Anlass, über die Gesundheit nachzudenken. Die Mehrheit der Menschen beeilt sich nicht, sich bei ähnlichen Fragen an Ärzte zu wenden: einige meinen, sie könnten Defekte selbständig bewältigen, andere schämen sich, sensible Probleme mit anderen zu teilen, und hoffen, dass die Beschwerden von selbst aufhören werden....
Abteilung: Artikel über Gesundheit
Fast jeder kennt sich mit Schnupfen aus und meint aufgrund seines eigenen Wissens und seiner Erfahrung, ihn richtig behandeln zu können.
Abteilung: Artikel über Gesundheit
Jede Frau hat Präferenzen bei der Nutzung von Waren, die ihr helfen, gut auszusehen, jung und wirkungsvoll zu fühlen; der Prozess der Auswahl des Lieblingsparfüms, Shampoos oder dekorativer Kosmetik beeinflusst diese Einstellung bereits...
Abteilung: Artikel über Gesundheit
Der Zustand der Lippen (ihre Verletzlichkeit und ihr Aussehen) ist ein wichtiges Kennzeichen des menschlichen Gesundheitszustands. Abschuppung, Trockenheit und Blässe sowie Risse in den Mundwinkeln können nicht nur auf kosmetische Mängel zurückzuführen sein, die durch physische Beschädigungen oder Wetterbedingungen entstanden sind, sondern auch auf Begleiterscheinungen bestimmter Erkrankungen und Organstörungen, die einer Behandlung bedürfen. Wir betrachten 10 mögliche Ursachen für das Auftreten von Mundwinkelrisse (Faulecken) in den Mundwinkeln sowie Wege zu ihrer Beseitigung...
Abteilung: Artikel über Gesundheit
Die Mehrheit der gynäkologischen Erkrankungen zeigt sich durch drei Hauptmerkmale, wobei jedes Merkmal auf die Notwendigkeit einer Diagnose hinweist...
Abteilung: Artikel über Gesundheit
Bierliebhaber in unserem Land sind sehr zahlreich. Nach Statistiken konsumiert jeder statistisch durchschnittliche Russe (einschließlich Frauen und Kinder) jährlich neben 60 Litern dieses Getränks. Dies ist nicht so viel wie in Tschechien oder Deutschland, doch die Zahl bleibt dennoch beeindruckend.
Abteilung: Artikel zur Gesundheit
Hyperorexie und Anorexie als schwerwiegende Abweichungen des Nahrungsverhaltens führen bei Patientinnen häufiger zum Tod als alle anderen neurotischen Störungen zusammen; in 60 % der Fälle begleiten sich beide Leiden: Die Patienten fürchten die Gefahr einer Gewichtszunahme und versuchen, auf Nahrung zu verzichten, leiden jedoch periodisch an plötzlichen Hungerattacken und unkontrolliertem Essen. Jedem Kranken mit Anorexie und Hyperorexie wird qualifizierte Hilfe empfohlen.
Abteilung: Artikel zur Gesundheit
Es scheint zunächst so, als gäbe es keine Differenzen: Wasser ist für den menschlichen Organismus unverzichtbar...
Abteilung: Artikel zur Gesundheit
Bei hohem Fieber – einem häufigen Symptom verbreiteter Erkrankungen wie ORVI, Angina oder Pneumonie –, um die Hitze zu senken und den Zustand des Kranken zu erleichtern, empfehlen Ärzte Antipyretika; jedoch ist ihre Anwendung nicht immer möglich. Zu früh...
Abteilung: Artikel zur Gesundheit
Varikose ist laut Statistik vielen bekannt; an dieser Erkrankung leiden etwa die Hälfte der gesamten erwachsenen Bevölkerung. In der Regel betrifft die Varikoze vorwiegend oberflächliche Venen und zeigt sich durch charakteristische kosmetische Defekte. Viel gefährlicher wird eine Thrombose tieferer Venen eingeschätzt, da dieses Leiden in frühen Stadien unmerklich verlaufen kann und bei fortgeschrittenen Fällen ernste Risiken birgt – die Thrombose. Dieser Zustand entsteht, wenn sich ein Blutpfropf bildet...
Abteilung: Artikel zur Gesundheit
Jeder Mensch weiß, dass eine Erhöhung der Körpertemperatur ein Merkmal von Unwohlsein ist; jedoch kann das Vorhandensein einer Erkrankung auch ohne Fieber auftreten...
Abteilung: Artikel über Gesundheit
Störungen des Immunsystems manifestieren sich häufig als Allergien; laut Statistik leiden mehr als 40 % der Weltbevölkerung daran, meist ausgelöst durch pathologische Reaktionen auf Lebensmittelzutaten, tierische Wolle oder Medikamente.
Abteilung: Artikel über Gesundheit
Über den Nutzen von Haustieren für die kindliche Entwicklung wurde bereits viel geschrieben. Dennoch zögern viele Eltern bei der Einführung neuer Tiere, da sie befürchten, die Kindergesundheit zu gefährden. Welche Risiken lauern wirklich auf die Kleinen und wie lässt sich das Zusammenleben mit Haustieren komfortabel und sicher gestalten?
Abteilung: Artikel über Gesundheit
Unter Neurose versteht man eine Störung des Nervensystems, bei der Abweichungen im Ablauf höherer nervöser Prozesse beobachtet werden.
Abteilung: Artikel über Gesundheit
In der großen Auswahl an Parfüm- und Kosmetikprodukten heben sich heute Mittel mit antibakteriellen Komponenten besonders hervor. Diese Kategorie von Gelen, Shampoos, Seifen, Cremes, Lotionen und weiteren Erzeugnissen wird von den Herstellern oft als Allheilmittel positioniert.
Abteilung: Artikel über Gesundheit
Die Begriffe "Krankheit" und "Patientin" leiten sich nicht zufällig vom Wortstamm "Schmerz" ab. In der Regel beeinträchtigen die Symptome dieser Leiden das Leben des Patienten erheblich. Dennoch gibt es Ausnahmen: Einige Erkrankungen weisen Merkmale auf, die sogar positive Emotionen hervorrufen können. Leider sind die meisten solcher Zustände schwer und unheilbar.
Abteilung: Artikel über Gesundheit
Singen: Alle Menschen singen gerne; kleine Kinder beschäftigen sich beim Gesang ohne tiefe Überlegungen zur Melodie, während Erwachsene dies meist anders tun.
Abteilung: Artikel über Gesundheit
In modernen Geschäften findet man ein umfangreiches Sortiment an Früchten und Gemüse. Die Konsumenten haben sich daran gewöhnt, dass auf den Ladentischen nicht nur einheimische Saisonprodukte, sondern auch Importfrüchte und -gemüse ganzjährig verfügbar sind.
Abteilung: Artikel über Gesundheit
Das Baden in Suds mit Heilkräutern (Phyto-Bäder) war bereits seit der Zeit Kleopatras verbreitet. Heute ist das Phyto-Bad ein einfaches und verfügbares Mittel, das nicht nur nervöse Anstrengungen lindert, sondern auch zur Behandlung vieler Erkrankungen beiträgt. Kräuterbäder sind bei der Therapie von Erkältungen, Osteochondrose, Radikulopathie, Hauterkrankungen sowie Gelenkbeschwerden besonders wirksam.
Abteilung: Artikel über Gesundheit
Eine reaktive Pankreatitis ist eine Entzündung der Bauchspeicheldrüse, die häufiger auftritt als andere Formen.
Abteilung: Artikel über Gesundheit
Wahrscheinlich gibt es keinen Menschen, dem eine Erkältung nicht wehtun würde; Schnupfen, Husten und Kopfschmerzen sind jedem bekannt. Im Herbst steigt die Zahl der Infektionen mit respiratorischen Viren: Schulen und Kindergärten werden betroffen, während Influenza langsam in die Städte vordringt.
Abteilung: Artikel zur Gesundheit
Das trophische Geschwür ist keine eigenständige Erkrankung, sondern eine schwere Komplikation, die durch thermische Traumata (Verbrennungen), langdauernde Pathologien der Adern oder Venen der unteren Extremitäten, Diabetes mellitus sowie bestimmte Infektionen des Bindegewebes, Lymphgefäße, Haut oder Nervenstämme ausgelöst wird. Die Pathologie manifestiert sich als nicht heilende Wunde an der medialen Oberfläche des Unterschenkels, auf der Fußsohle, am Fersenbereich oder an den Zehen.
Abteilung: Artikel zur Gesundheit
Die extrakorporale Befruchtung ist eine der modernsten Methoden zur Bekämpfung von Unfruchtbarkeit. Bis heute haben bereits viele Menschen davon Nutzen.
Abteilung: Artikel zur Gesundheit
Vor 10 bis 15 Jahren galt das Vorhandensein eines Computers in einer Wohnung als Seltenheit, und die Büroräume waren lediglich auf dem ersten Stadium der Ausstattung mit diesen nützlichen Anlagen beschränkt. Heute ist ein Computer in fast jedem Haushalt üblich.
Abteilung: Artikel zur Gesundheit
Sphingolipidosen sind eine Gruppe von Erbkrankheiten, bei denen der Abbau bestimmter Lipide gestört ist. Dies führt zur Akkumulation von Sphingolipiden in verschiedenen Geweben, insbesondere im Zentralnervensystem und in den Knochenmarkzellen.
Abteilung: Artikel zur Gesundheit